

Abstract
Germline SAMD9L mutation is a recently recognized cause of constitutional bone marrow failure with a unique propensity for clonal evolution to myelodysplastic syndrome (MDS) with monosomy 7. It is now known that a clinical syndrome originally described in the 1970s as Ataxia-Pancytopenia Syndrome is caused by missense mutation in SAMD9L, and subsequent case series have demonstrated diverse clinical outcomes in patients with germline SAMD9L mutations. SAMD9L is located on chromosome 7, and gain-of-function mutations in SAMD9L is thought to decrease cell proliferation by inhibiting the normal cell cycle. In patients with SAMD9L mutations, the development of a monosomy 7 clone then rescues the inhibitory effect of mutant SAMD9L but can result in progression to acute leukemia. Interestingly, there are also reports of transient monosomy 7 due to uniparental disomy occurring in the monosomy 7 clone resulting in normal hematopoiesis.
We describe the case of a male patient who presented at age 8-months of age with fever, hypotension, and pancytopenia. Bone marrow biopsy was hypocellular with debris-laden histiocytes. Diagnostic evaluation included normal chromosome stability testing (DEB and MMC), normal telomere length testing, and normal gene panels for inherited bone marrow failure and primary HLH. Patient, then, had spontaneous count recovery on day 41 of illness with progressive improvement becoming transfusion independent without any further infections. Repeat bone marrow biopsy 1 month after count recovery demonstrated dysplasia and cytogenetic evaluation revealed monosomy 7 in 11 out of 20 metaphases. Whole exome sequencing demonstrated a novel mutation in SAMD9L. Parents were analyzed for the SAMD9L mutation which were negative. Repeat bone marrow evaluation at 4, 7, and 10 months after presentation continued to demonstrate dysplasia and monosomy 7. After 12 months of observation, decision was made to perform a bone marrow transplant but the patient died from diffuse alveolar hemorrhage shortly after engraftment.
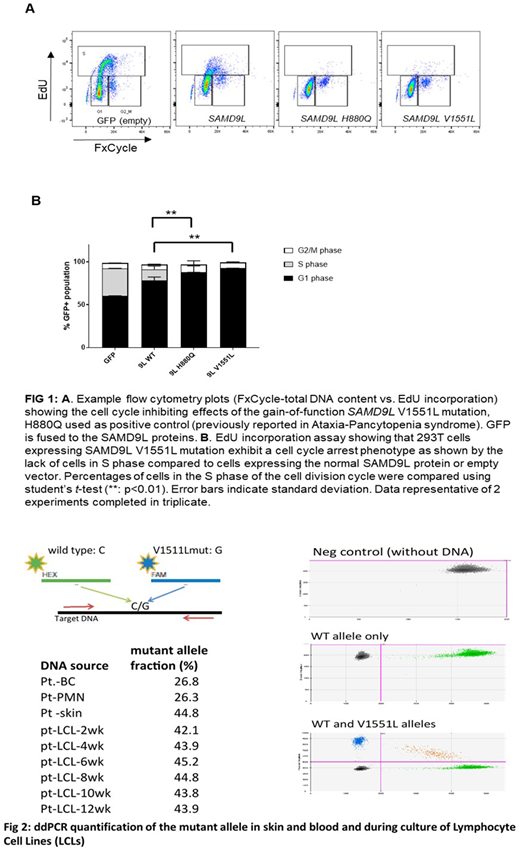
EdU incorporation assay showed that 293T cells expressing SAMD9L V1551L mutation exhibit a cell cycle arrest phenotype as shown by the lack of cells in S phase compared to cells expressing the normal SAMD9L protein or empty vector. Furthermore, Digital droplet PCR (ddPCR) with wild type- and mutation-specific primers was used to quantify the percent of mutant allele in the patients' fibroblasts, whole blood, granulocytes, lymphocytes and lymphocyte cell cultures over 12 weeks. The patient's whole blood DNA had a low fraction of the mutation (26.8%), supporting the cytogenetic finding of mosaic monosomy 7. Interestingly, lymphocyte cell line generated from the patient's blood showed a higher fraction of mutation in about 40-45% similar to what is seen in fibroblasts which suggests that the patient's monosomy 7 clone which rescued the neutrophil count seems to preferentially contribute to the myeloid lineage versus the lymphocyte lineage. We have further developed induced pluripotent stem cells from this patient to further explore this question.
In summary, our case describes a novel SAMD9L heterozygous missense mutation (p.Val1551Leu) resulting in gain-of-function mutation which inhibits cellular proliferation resulting in bone marrow failure. This case highlights the new insights offered through the use of whole exome sequencing and also the difficulty of making the clinical decision to treat with bone marrow transplant in the setting of multiple reports of disease remission via self-correction. If we are able to identify the clone giving rise to mature cells and quantify it over time, we may be able to provide a more informed recommendation regarding bone marrow transplant versus observation.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal